 | Abbreviations Key |
| CITE-seq | cellular indexing of transcriptomes and epitopes sequencing |
| HISE | Human Immune System Explorer |
| IDE | integrated development environment |
| NPX | normalized protein expression |
| PEA | proximity extension assay |
| QC | quality control |
| scRNA-seq | small cytoplasmic RNA sequencing |
| scVI | single-cell variational inference |
| UMAP | uniform manifold approximation and projection |
| VM | virtual machine |
Manage NextGen IDE Packages
At a Glance
HISE NextGen IDEs are scalable, secure, version-controlled yet temporary workspaces in which to conduct experiments and analyze data. This document outlines the folder structure and workflow of HISE NextGen IDEs and explains how to install, enable, or disable Jupyter extensions. If you have questions or suggestions, contact immunology-support@alleninstitute.org.
Description
When a new HISE IDE is provisioned, a virtual machine (VM) is created. This environment contains tools and libraries defined by key scientists based on specified project needs, such as scRNA-seq or flow cytometry. To keep your workspace lightweight and speed up package availability, the VM loads only what you need. To perform even more specialized tasks, you can install Jupyter Notebook extensions.
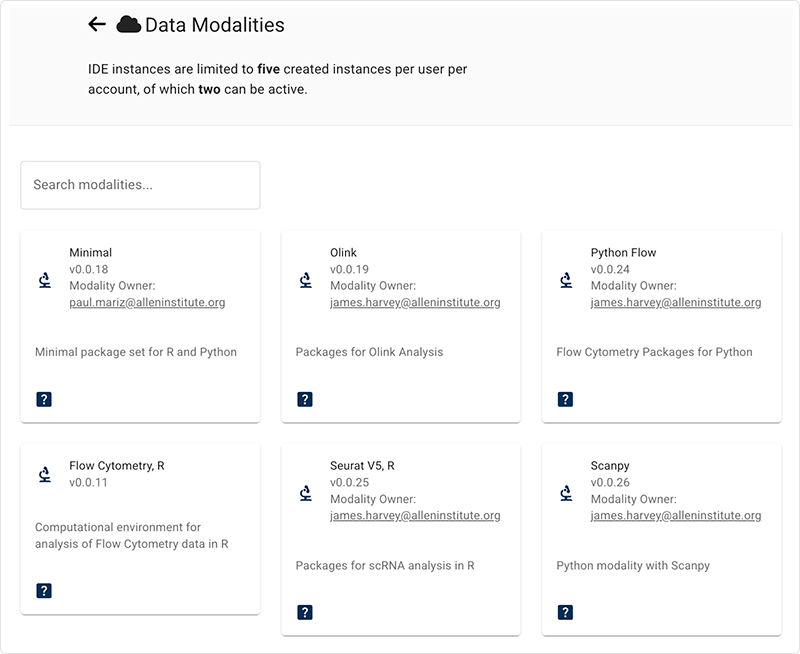
Data Modalities
NextGen IDEs are designed to work with specific data modalities, outlined in the following table:
| Data modality | Designed for | Examples of data yielded |
| R_seurat_v5 | scRNA-seq analysis with the Seurat package in R | Large-scale gene expression data, UMAP projections, cell type annotation |
| olink | Olink processing and protein analysis of small biological samples using PEA technology | NPX data, QC data, biomarker studies |
| python_flow | Flow cytometry data analysis in Python | Single-cell protein expression data, cell population frequencies, cell phenotypes |
| python_scvi | Single-cell genomics analysis using the scVI package in Python | Differential gene expression, CITE-seq data, cell annotation, spatial transcriptomics |
| r_flow | Flow cytometry data analysis in R | Single-cell protein expression data, cell population frequencies, cell phenotypes |
When you create a new IDE, you choose your preferred modality, as shown in the following screenshot:
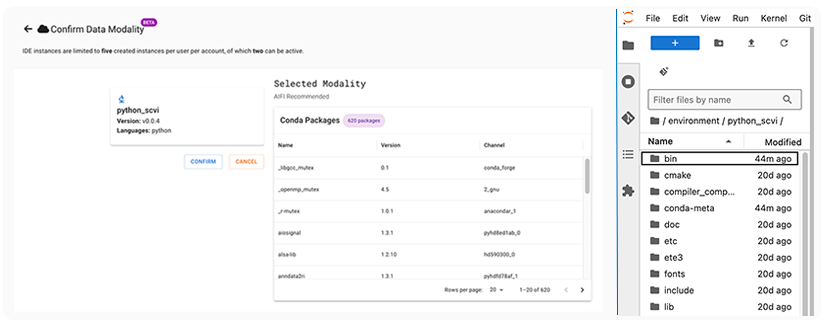
The tools for the selected modality load automatically. You can see them either in HISE, before you confirm your modality choice (see below, left), or in the ~/environment folder of any IDE notebook created from the modality (see below, right). The packages are updated as needed. For example, in the environmment folder for the python_scvi modality, you can see that the /bin folder was last modified 44 minutes before this screenshot was captured.
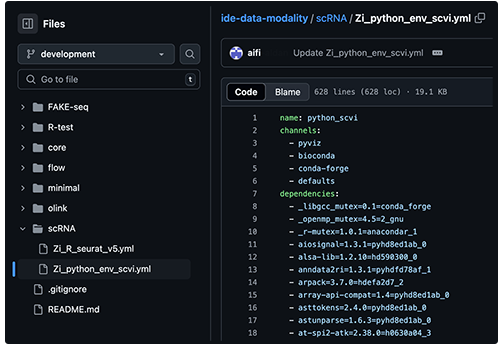
Data modality tools are chosen and maintained by experts in the selected field. They're stored in a dedicated Git repository like the scRNA repo shown here:
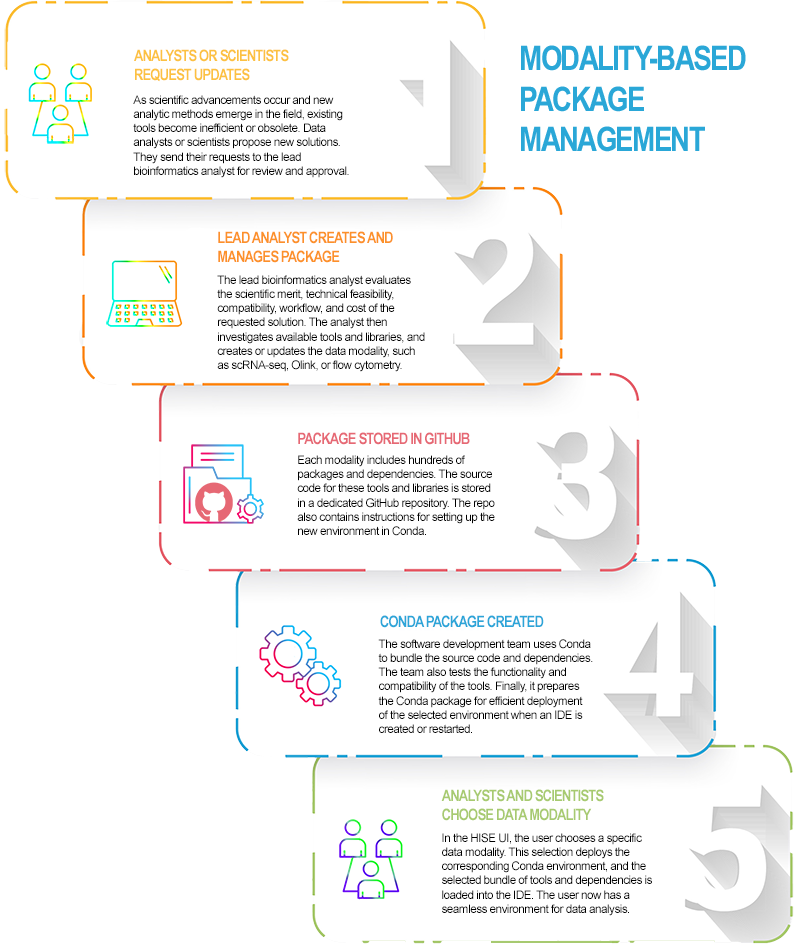
Each modality contains hundreds of packages. If the selected IDE configuration doesn't meet your needs, however, you can customize it by adding Jupyter extensions. For details, see the IDE Extensions section of this document. The following image summarizes the modality-based package management process.
IDE Extensions
JupyterLab supplies preconfigured extensions you can install with Python pip or conda packages.
List extensions
To list all installed Jupyter extensions, enter the following command in your terminal:
jupyter labextension list
Disable extensions
Jupyter extensions are enabled by default. To disable a Jupyter extension, enter the following command in your terminal:
jupyter labextension disable my-extension
Enable extensions
To enable a Jupyter extension, enter the following command in your terminal:
jupyter labextension enable my-extension
 Related Resources
Related Resources
Create Your First NextGen IDE Instance (Tutorial)